|
|
とうやく396号(2013年1月号)学術欄HOME > 学術欄 |
|

痛風・高尿酸血症研究における最近の進歩
はじめに
尿酸はプリン体の最終代謝産物であり、血清尿酸値は尿酸への代謝量(産生量)と腎臓を中心とした尿酸排泄能のバランスにより規定されていると考えられてきた。 尿酸は細胞膜を自由に通過することができないため、トランスポーターを介して輸送される。したがって、尿酸の排泄は尿酸トランスポーターにより制御されているといえる。 最近、トランスポーター研究を通して、高尿酸血症の発症メカニズムについて多くの知見が集積されてきた。 これまで高尿酸血症の発症機序として尿酸産生過剰と腎臓からの尿酸排泄低下の2つが考えられていたが、今回、尿酸トランスポーターであるABCG2の研究から、新たに尿酸の腎外排泄低下による高尿酸血症も存在することが明らかになった。 本稿では、この新しい概念である腎外排泄低下型高尿酸血症について概説する。 A 高尿酸血症の病型分類 高尿酸血症は、尿酸排泄低下型、尿酸産生過剰型、そして両者の機序を持つ混合型に分類されてきた。 高尿酸血症の中で、尿酸排泄低下型の頻度が55-60%、尿酸産生過剰型が10-15%、そして混合型が25-30%といわれている(図1)。 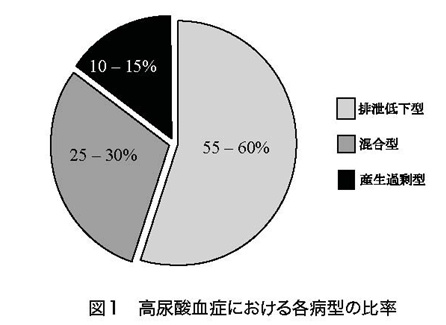
混合型を含めて考えると尿酸排泄低下の機序による高尿酸血症は全体の85-90%で、同様に尿酸産生過剰の機序によるものが40-45%になる。 したがって、ほとんどの高尿酸血症は尿酸の排泄低下が関係していると同時に、その半数近くは尿酸の産生過剰が関係しているとも言える。 この尿酸排泄低下の機序としては、尿酸トランスポーター遺伝子の一塩基置換などによる尿酸輸送能の差が原因と推定されている。尿酸産生過剰の成因としての遺伝の関与については、Lesch-Nyhan症候群など頻度の著しく低い先天性代謝異常症が知られているものの、それら以外は不明であり、大多数の尿酸産生過剰はプリン体過剰摂取などの生活習慣の影響が大きいと考えられてきた。 B 尿酸動態 通常、ヒトの体内尿酸プールは、成人男性で約700-1700mg(平均1200mg)であり、成人女性は550-700mgといわれている(図2)。 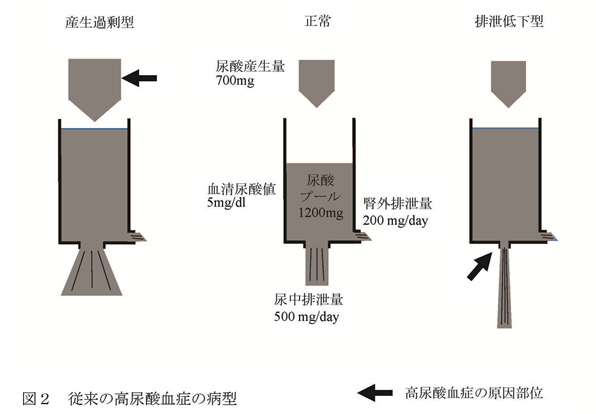
食事、生体内のプリン体合成や細胞の分解の結果、1日あたり700-800mgのプリン体が尿酸プールに流入するが、同量の尿酸が体外に排泄されることにより、血清尿酸値はほぼ一定に保たれている。尿酸の約2/3は腎臓から、残りのほとんどは消化管から排泄される。 腎臓において尿酸は、糸球体濾過膜を自由に通過した後、尿酸トランスポーターを介して再吸収と分泌が両方向性に行われ、最終的には糸球体を通過した尿酸の6ー10%が尿中に排泄される。 高尿酸血症のほとんどが腎臓の尿酸排泄低下によること、体外への尿酸排泄の約2/3が腎臓から行われていること、そして多くの物質の血清レベルが腎臓におけるトランスポーターの制御により一定に保たれていることから、血清尿酸値は腎臓からの排泄により制御されていると考えられてきた。 C 尿酸トランスポーターABCG2 腎臓における尿酸輸送は主に近位尿細管で行われるが、現在までにURAT1を含む、いくつかの尿酸トランスポーターが同定されている1(図3)。 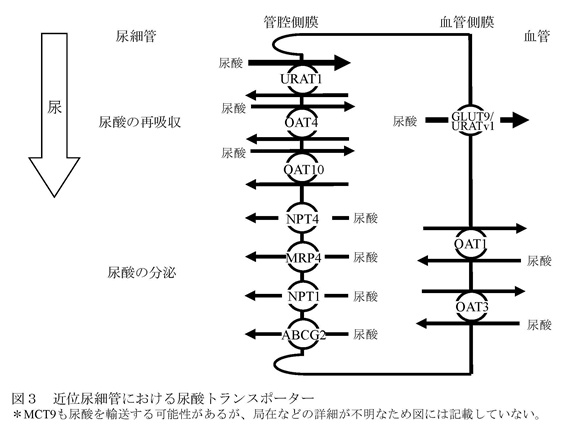
この中で、尿酸の再吸収に主に関与しているのが、近位尿細管の管腔側膜のURAT1と血管側膜のGLUT9/ URATv1である。一方、尿酸の分泌に主に関与しているのが、ABCG2である。 ABCトランスポーターは、ATP結合カセット(ATP-Binding Cassette)と呼ばれる共通配列を有し、ATPを利用して物質の能動輸送を行い、細胞内から外へ物質を汲み出す排泄ポンプとして働く。 主にABCG2は、小腸、肝臓、腎尿細管等の管腔側膜に発現し、有機アニオン系化合物の排泄を行っている。このABCトランスポーターであるABCG2の遺伝子は、当初薬剤耐性に関連する遺伝子として、薬剤耐性を示す乳がん細胞からクローニングされた。 2004年に痛風の関連遺伝子の候補領域が第4染色体長腕に存在することが報告されたことから、領域内のトランスポーターについての検索及び解析が始まった2。 その後、関連する遺伝子を網羅的に抽出する手法である全ゲノム関連解析の論文において、血清尿酸値と関連性を持つ分子のデータの中に第4染色体長腕に存在するABCG2も載った3。 我々は、直ちにABCG2にターゲットを絞り、ABCG2の尿酸輸送能の検討及び痛風患者を用いた遺伝子解析を行った。HEK293細胞にABCG2を発現させた細胞膜小胞を調製して尿酸の輸送能を解析した結果、ABCG2が尿酸を輸送すること、また、生理的な尿酸濃度範囲において、輸送飽和を生じない、すなわち頭打ちになることなく尿酸輸送を増加させる高容量性の尿酸トランスポーターであることが明らかになった。また、ABCG2の機能低下が痛風の発症に関連していることも判明した。これらの知見は、我々を含む2つのグループが同時に報告した4,5。 日本人のABCG2の遺伝子変異の中で、126番目のグルタミンが終止コドンになるQ126Xと141番目のグルタミンがリシンになるQ141Kのアレル頻度は、それぞれ約3%と32%で、高頻度に認められる6。 変異体を用いた機能解析の結果、Q126Xでは尿酸輸送能が消失するのに対し、Q141Kでは輸送能が半分に減少することが判明した5。 また、2つの一塩基多型は、1つの染色体上には同時に存在しないことから、独立したリスクとして計算できることが明らかになった5。 そこで、これを用いて、159例の痛風患者のABCG2の機能を評価した。その結果、ABCG2の機能低下にしたがい痛風の発症リスクは上昇し、機能が4分の1以下になる遺伝子変異を持つ患者では、正常例に比し痛風の発症リスクが約26倍にも上昇することが判明した(表1)5。 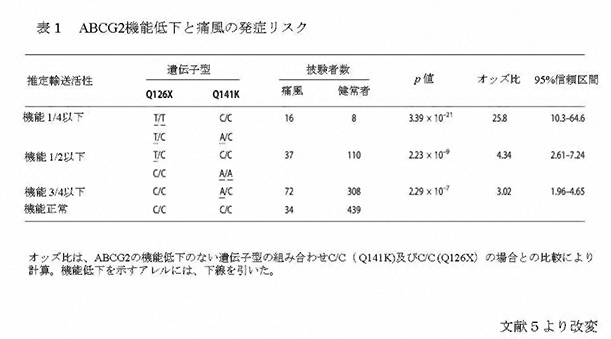
このABCG2の機能変化は、痛風・高尿酸血症患者だけでなく、一般健常者の血清尿酸値にも影響を与え、日本人の健康診断受診者739例において、Q141Kの変異数が多いほど血清尿酸値が上昇していた(図4)。 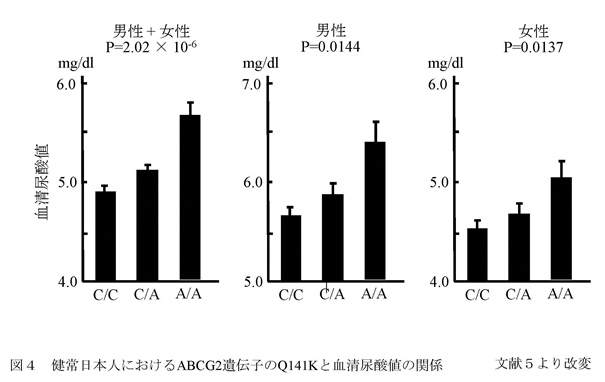
これらの結果から、2つのABCG2の一塩基多型が血清尿酸値に影響を与え、痛風・高尿酸血症発症に大きく関与していることが明らかになった。 腎外排泄低下型高尿酸血症 ABCG2は、他の尿酸トランスポーターと異なり腸管など多くの臓器に発現し、腎臓にも発現していることから、腎臓ABCG2の機能低下により、尿中への尿酸排泄が低下し、高尿酸血症を引き起こすことが予想されたが、詳細は不明であった。 そこで、ABCG2による高尿酸血症の発症機序を明らかにするために、644人の高尿酸血症患者を対象として、ABCG2の機能低下と腎臓の尿酸排泄との関係を検討した7。 その結果、予想に反して、ABCG2の尿酸排泄能が低下していると、腎臓からの尿酸排泄量は増加していることが明らかとなった。 また、ABCG2の尿酸排泄機能の低下は、高尿酸血症患者の約8割(76%)に認められた。 さらに、ABCG2機能正常高尿酸血症に対するABCG2機能低下高尿酸血症の尿酸産生過剰をきたす高尿酸血症(尿酸産生過剰型+混合型)の相対危険度は、最大で約2.3であった。 つまり、ABCG2の機能が低下すると、従来の分類における尿酸産生過剰型や混合型になりやすいことが判明した。 これは、ABCG2が腸管にも発現していることから、腸管における尿酸排泄が低下し、そのため高尿酸血症を来すことが推定された。 もちろん、腎臓のABCG2を介した尿酸排泄も低下するが、腎臓には他の尿酸トランスポーターも存在し、それを介して尿酸が排泄され尿中尿酸排泄量が増加する。 しかし、これだけでは腸管からの尿酸排泄減少をカバーしきれず、高尿酸血症を来すものと考えられた。この仮説を証明するためには、腸管に排泄された尿酸量の測定が必要であるが、腸管では常在細菌叢により尿酸が分解されるため尿酸量の定量ができない。 そのため、abcg2ノックアウトマウスを作成し、尿酸代謝酵素であるウリカーゼ阻害薬であるオキソン酸を投与し、尿酸排泄について検討を行った。 その結果、ノックアウトマウスでは、ABCG2機能が低下した高尿酸血症患者と同様に、血清尿酸値と腎臓からの尿酸排泄が増加しただけでなく、腸管からの尿酸排泄の低下が観察された。 これにより、前述の高尿酸血症発症のメカニズムがヒトにおいても起こりうることが示された(図5)。 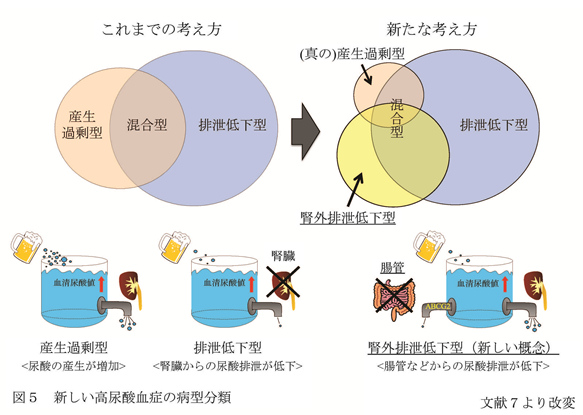
従来の高尿酸血症の病型分類においては、尿中への尿酸排泄量等を規準としているため、尿酸が尿中に多く排泄されることは、尿酸が体内で多く産生される尿酸産生過剰型または混合型高尿酸血症を意味していた。 しかし、今回の結果は、腎臓以外(腸管)からの尿酸排泄の低下によっても、従来の分類における尿酸産生過剰型または混合型高尿酸血症を呈することを示している。 これは、今まで無視されがちであった腸管からの尿酸排泄の重要性を示すものであり、このメカニズムは、多くの高尿酸血症発症に関与している。 以上のことから、高尿酸血症の原因として新たに腎外排泄低下型高尿酸血症という概念を提示し、腎外排泄低下型高尿酸血症と真の産生過剰型高尿酸血症をあわせた、従来の尿酸産生過剰型高尿酸血症を、腎負荷型高尿酸血症と改名することをあわせて提唱した。 参考文献
|
|